Desórdenes genéticos
Síndrome de Angelman

El síndrome de Angelman es un desorden neurológico, asociado a un retraso mental. Durante varias décadas el estudio del cromosoma de los individuos con Angelman no reveló ninguna anormalidad, pero con el desarrollo de mejores métodos de análisis, se encontró que en el cromosoma 15 faltaba un área muy pequeña.
La hiperactividad y la sonrisa frecuente son probablemente las conductas más típicas en el SA junto a las convulsiones en los casos más graves y los trastornos de alimentación. Todo esto hace que los niños con SA deban recibir un completo abanico de entrenamiento temprano y programas de estimulación. En 1965 el médico inglés Harry Angelman, describió por primera vez, al observar similares características en tres niños, lo que hoy conocemos como el Síndrome de Angelman (SA). Dentro de estas características el Dr. Angelman identificó que los tres niños tenían rigidez, andar espástico, ausencia de habla, risa excesiva y crisis convulsivas.
El siguiente es un extracto de las anotaciones de Angelman respecto a su descubrimiento:
“La historia de la medicina está llena de historias interesantes sobre el descubrimiento de enfermedades. La saga del síndrome de Angelman es una de esas historias. Fue por pura casualidad que hace casi treinta años, tres niños discapacitados fueron ingresados en varias ocasiones en mi unidad de pediatría en Inglaterra. Tenían diferentes discapacidades y aunque a primera vista parecían estar padeciendo afecciones diferentes, yo tenía la sensación que había una causa común para su enfermedad. El diagnóstico fue puramente clínico, porque a pesar de testearlos con las pruebas físicas existentes, las cuales hoy en día son mucho más refinadas, no fui capaz de establecer con una prueba científica que los tres niños tenían la misma discapacidad. En vista de esto dudé publicar mis estudios sobre ellos en revistas médicas. Sin embargo, estando de vacaciones en Italia, vi una pintura al óleo en el museo de Castelvecchio en Verona llamada “Un muchacho con una muñeca”. La cara de sonrisa del muchacho y el hecho de que mis pacientes mostraran movimientos rígidos me dieron la idea de escribir un artículo sobre los tres niños con el título de ‘Niños muñeca’ (happy puppets, o también muñecos o títeres felices). No fue un nombre que agradara a todos los padres pero sirvió como un medio de incluir a los tres pequeños pacientes en un solo grupo. Después el nombre se cambió al de síndrome de Angelman. Este artículo se publicó en 1965 y después de un cierto interés inicial casi se olvidó hasta principios de los años ochenta”.
Una de las causas que demoró un diagnóstico preciso es que el síndrome de Angelman, por lo general, no se reconoce en el recién nacido o en la infancia, dado que los problemas de desarrollo no son específicos durante este tiempo.
Incluso los padres suelen sospechar el diagnóstico después de haberse informado sobre el SA o ver a otro niño que reúna esas características.
Por esto mismo, en la mayoría de los casos el diagnóstico de SA se realiza entre los tres y los siete años, cuando las conductas características y los rasgos se hacen más evidentes.
Todos los rasgos no necesitan estar presentes para que el diagnóstico pueda ser hecho y éste, a menudo, es lo primero que se sospecha cuando las conductas típicas son reconocidas.
Incidencia y características principales
Hoy sabemos que el síndrome de Angelman es un desorden genético. Estudios de estas características han identificado pequeñas tachaduras en el cromosoma 15 en aproximadamente 80% de los individuos con el síndrome de Angelman. En un subgrupo muy pequeño que recibieron dos cromosomas 15 normales del papá, se presentó el síndrome de Angelman. En el 20% restante, los estudios genéticos fueron negativos y el diagnóstico se mantuvo como caso clínico.
En 1997 se identificó un gen (UBE3A) en el síndrome de Angelman y recientemente han sido encontradas mutaciones en niños con este síndrome que antes habían sido diagnosticados como casos clínicos.
Se han detectado casos de síndrome de Angelman en todo el mundo, en diferentes grupos raciales y afecta a hombres y mujeres por igual. En Estados Unidos la gran mayoría de los casos conocidos parecen ser de origen caucásico. Su incidencia varía entre distintos informes que reportan estimaciones comprendidas de 1 caso por cada 15.000/30.000 ó 1 por cada 12.000/20.000 personas.
Las características generales predominantes del SA son:
Siempre (100% de los casos):
- Retraso en el desarrollo, funcionalmente severo.
- Capacidad de habla, ninguna o uso mínimo de palabras; las habilidades de comunicación receptivas y no verbales mayores que las verbales.
- Problemas de movimiento y de equilibrio, normalmente ataxia al andar y/o movimiento trémulo de miembros.
- Conducta característica y singular: cualquier combinación de risa/sonrisa frecuente; apariencia de felicidad; personalidad fácilmente excitable, a menudo con movimientos de aleteo de manos; hipermotricidad; permanencia de la atención durante poco tiempo.
Frecuente (más del 80%):
- Retraso, crecimiento inferior al normal del perímetro cefálico, normalmente produciendo microcefalia (absoluta o relativa) alrededor de los 2 años de edad.
- Crisis convulsivas normalmente antes de los 3 años de edad.
- Electroencefalograma (EEG) anormal, modelo característico con ondas de gran amplitud y picos lentos.
Asociado (20 - 80%):
- Estrabismo.
- Hipopigmentación de piel y ojos.
- Lengua prominente; problemas para succionar y tragar.
- Reflejos de tendones hiperactivos.
- Problemas de alimentación durante la infancia.
- Brazos levantados y flexionados al andar.
- Mandíbula prominente.
- Hipersensibilidad al calor.
- Boca ancha, dientes espaciados.
- Problemas para dormir.
- Babeo frecuente.
- Lengua prominente.
- Atracción hasta la fascinación por el agua.
- Conductas excesivas de mascar/masticar.
- Achatamiento de nuca.
Características del lenguaje del niño con síndrome de Angelman:
- Falta de lenguaje oral; la mayoría de ellos suelen decir pocas palabras, y sin sentido comunicativo.
- El nivel comprensivo está algo más preservado y la memoria para recordar las caras es buena.
- Las conductas lingüísticas pre-verbales también están muy alteradas, no suelen hacer juego vocal ni gestos naturales.
- Existe dificultad en señalar con el dedo.
- En algunos casos, utilizan sistemas de comunicación alternativa con éxito.
Diagnóstico
El diagnóstico del SA se realiza a través de criterios clínicos y de pruebas de laboratorio. Mediante el estudio de genética molecular se detecta la pérdida de la contribución materna a la región AS/PWS (locus cromosómico 15q11-q13) que se produce en el 80% de los casos. El resto de pacientes (20%) que presentan criterios clínicos de síndrome de Angelman pero con pruebas genéticas normales, probablemente presentan una mutación en el gen UBE3A.
La prueba genética más exacta es el test de fluorescencia in situ hibridación (F.I.S.H).
Es a través de estudios como éste que el área anulada (las pequeñas tachaduras en el cromosoma 15), aunque sumamente pequeña, es realmente bastante grande cuando se analiza a nivel molecular.
Se sabe que la región anulada en el cromosoma 15 contiene genes que están activados o desactivados, dependiendo del origen materno o paterno del cromosoma (por ejemplo, un gen puede estar activado en el cromosoma 15 heredado de la madre pero el mismo gen heredado del padre puede que esté desactivado). Este mecanismo de activación específica parental es conocido como “imprinting” genético. Dado que las deleciones vistas en SA sólo ocurren en el cromosoma 15 heredado de la madre, se pensó que el gen o genes responsable del SA sólo se activaba en el cromosoma materno. La deleción en genes que están activados y que son de origen paterno causa otro desorden de retraso mental conocido como el síndrome de Prader-Willi (SPW). Los genes de SPW se localizan cerca del gen SA, pero son diferentes.
 En 1997 se vio que un gen localizado en la región delecionada de SA llamado UBE3A podía estar mutado en aproximadamente un 5% de los individuos de SA (22,23). Estas mutaciones pueden ser tan pequeñas como un par de bases. El gen codifica una proteína llamada “Ubiquitin Protein Ligase” y se cree que el UBE3A puede ser el gen causante de SA. Todos los mecanismos conocidos que causan SA presentan o provocan la inactivación o ausencia de este gen. UBE3A es un componente encimático de un complejo sistema de degradación de la proteína llamada “Ubiquitin-Proteasome pathway (camino)”. Este camino está localizado en el citoplasma de todas las células. El camino implica a una pequeña molécula, Ubiquitin, que puede estar unida a proteínas y de ese modo provocar que éstas sean degradadas (24). En un cerebro normal, la copia del UBE3A heredado del padre está casi completamente inactiva; por tanto, la copia materna desempeña la mayor parte de las funciones del UBE3A en el cerebro. Las mutaciones del UBE3A heredadas de la madre causan el SA; las mutaciones del UBE3A heredadas del padre no tienen efectos detectables en el niño. En algunas familias, el SA causado por mutación del UBE3A puede darse en más de un miembro.
En 1997 se vio que un gen localizado en la región delecionada de SA llamado UBE3A podía estar mutado en aproximadamente un 5% de los individuos de SA (22,23). Estas mutaciones pueden ser tan pequeñas como un par de bases. El gen codifica una proteína llamada “Ubiquitin Protein Ligase” y se cree que el UBE3A puede ser el gen causante de SA. Todos los mecanismos conocidos que causan SA presentan o provocan la inactivación o ausencia de este gen. UBE3A es un componente encimático de un complejo sistema de degradación de la proteína llamada “Ubiquitin-Proteasome pathway (camino)”. Este camino está localizado en el citoplasma de todas las células. El camino implica a una pequeña molécula, Ubiquitin, que puede estar unida a proteínas y de ese modo provocar que éstas sean degradadas (24). En un cerebro normal, la copia del UBE3A heredado del padre está casi completamente inactiva; por tanto, la copia materna desempeña la mayor parte de las funciones del UBE3A en el cerebro. Las mutaciones del UBE3A heredadas de la madre causan el SA; las mutaciones del UBE3A heredadas del padre no tienen efectos detectables en el niño. En algunas familias, el SA causado por mutación del UBE3A puede darse en más de un miembro.
Otra causa del SA (2,3% de los casos) es la Disomía Uniparental (UPD), donde el niño hereda del padre ambas copias del cromosoma 15, y ninguna copia es heredada de la madre. En este caso, no hay deleción o mutación pero el niño no tiene el gen UBE3A activado, ya que los cromosomas de origen paterno sólo tienen genes UBE3A inactivos.
Un cuarto tipo de individuos con SA (3-5% de los casos) han heredado copias del cromosoma 15 del padre y de la madre, pero la copia heredada de la madre funciona de la misma forma que la copia paterna, es decir, está inactivo. Esto es denominado como un “defecto en el imprinting”. Algunos individuos con SA con defecto en el imprinting tienen una deleción muy pequeña de la región llamada centro del imprinting (IC) (25,26). El IC parece ser capaz de ejercer su efecto en UBE3A desde una localización lejana pero no se sabe todavía cómo se produce este mecanismo de actuación. En algunos casos, el SA causado por defectos en el imprinting puede afectar a más de un miembro de la familia.
Estos descubrimientos han llevado a saber que existen varios “tipos” de mecanismos genéticos que producen SA (25,27).Todos estos mecanismos, generalmente, conducen a las típicas características clínicas observadas en el SA; no obstante, se pueden producir pequeñas diferencias entre distintos grupos.
Convulsiones e hiperactividad
Como se ha destacado en las principales características y variando entre distintos estudios, el 80 - 90% de los portadores de SA han manifestado convulsiones. Por otra parte, los rasgos de hiperactividad son casi un denominador común a todos los casos.
En el punto que atañe a las convulsiones, cabe destacar que puede tratarse de una sobrestimación, pues los informes médicos tienden a estudiar los casos más severos. Menos del 25% de los niños afectados con SA padecen convulsiones antes de los 12 meses de edad. La mayoría tiene convulsiones antes de los 3 años; la incidencia en niños mayores o en adolescentes no es excepcional.
Las convulsiones pueden ser de cualquier tipo:
- De tipo motor, afectando a todo el cuerpo con sacudidas de las extremidades.
- De tipo ausencia, que conllevan periodos breves de falta de consciencia y que pueden requerir medicaciones múltiples anticonvulsivas.
Las convulsiones pueden ser difíciles de reconocer o diferenciar de temblores normales del niño, movimientos hiperquinéticos de extremidades o faltas de atención. El electroencefalograma típico, es a menudo más anormal de lo esperado y puede hacer pensar en actividad convulsiva cuando, de hecho, no la hay.
No hay consenso acerca de la medicación anticonvulsiva óptima, no obstante el ácido valproico (Depakine), topiramate (Topamax), carbamacepina (Tegretol), clonacepam (Klonopin), ethosuximide (Zarontin), son prescritos más comunmente que fenitoína (Dilantin), fenobarbital o ACTH (hormona adrenocorticotrópica). Es preferible el uso de medicación única pero es común que las crisis continúen. Algunos niños con convulsiones incontrolables han sido puestos en dieta cetónica, pero no está claro si esto es beneficioso. Los niños con SA tienen el riesgo de ser sobretratados con medicación porque pueden confundirse sus movimientos espásticos o faltas de atención con convulsiones, pueden dar EEG anormales incluso cuando las crisis convulsivas están controladas.
La hiperactividad probablemente es la conducta más típica en el SA. Se describe mejor como hipermotricidad con un bajo tiempo de atención. Durante la niñez o la adultez pueden tener una actividad aparentemente incesante, constantemente metiendo sus manos o juguetes en su boca, moviéndose de un sitio a otro. En casos extremos, el movimiento constante puede causar accidentes con contusiones y rozaduras. Conductas como agarrar, pellizcar y morder a niños más mayores se ha constatado que, también, pueden acentuarse por la actividad hipermotórica. Terapias persistentes y consistentes de modificación de conducta ayudan a disminuir o eliminar estas conductas no deseadas.
El tiempo de atención puede ser tan corto que impida la interacción social al no poder el niño con SA captar las expresiones faciales y otras señales sociales. En casos leves, la atención puede ser suficiente para aprender lenguaje de signos y otras técnicas de comunicación. Para estos niños, los programas de entrenamiento educativos y de desarrollo son fáciles de estructurar y generalmente son eficaces. Observaciones en jóvenes adultos sugieren que la hiperactividad disminuye con la edad. La mayoría de los niños con SA no toman medicación para la hiperactividad aunque algunos podrían beneficiarse del uso de medicaciones como methyiphenidate (Ritalina). El uso de agentes sedantes como phenothiazines no está recomendado debido a su potencia y efectos secundarios.
La risa en el síndrome de Angelman
Aunque hay un tipo de convulsión asociado con la risa, llamada epilepsia risible, esto no es lo que ocurre en el SA. La risa en el SA podría estar asociada a un suceso de expresión motora; la mayoría de las reacciones a los estímulos, físicos o mentales, se acompaña por risa o una sonrisa parecida a las muecas faciales. Aunque los niños con SA experimentan una variedad de emociones, aparentemente predomina la felicidad.
La primera evidencia de esta conducta característica puede estar en el comienzo de una temprana y persistente sonrisa a la edad de 1-3 meses. Risueños, sonriendo entre dientes y con sonrisa constante, pronto desarrollan una risa reflexiva normal, pero tienen retraso o están reducidas las conductas como arrullarse y parlotear. Más adelante varios tipos de expresiones faciales o conductuales caracterizan la personalidad del niño. Unos pocos presentan una risa verdaderamente cercana al paroxismo o contagiosa y en un estudio, el 70% de los casos presentó “estallidos de risa”.
La educación de los niños con SA
Se ha demostrado a través de muchos estudios que en los niños con SA que han recibido intervenciones educativas y de comportamiento se han alcanzado excelentes resultados en los niveles de comunicación, conducta social, educación y en los comportamientos relacionados a los hábitos de sueño y alimentación. Por otra parte, la fisioterapia y las intervenciones profesionales en el habla, sumados a modificaciones de conductas por parte de la familia, han aportado significativamente en el progreso de los niños y adolescentes.
También son de gran ayuda la terapia ocupacional (que puede ayudar a mejorar la motricidad fina y controlar las conductas motórico-bucales), la fonoaudiología, la terapia de comunicación y el entrenamiento en los métodos de comunicación no verbales.
Al ser los niños con SA sumamente activos, muchas veces será necesario equipar especialmente el aula y pueden necesitarse profesores de apoyo para integrar al niño en el aula. Por ello, la distribución del espacio debe estructurarse tanto en el plano físico como en su programa de actividades, para que el niño hiperactivo pueda ocupar su lugar y adaptarse al ambiente escolar. La individualización y flexibilidad son factores importantes. Técnicas de modificación de conducta, tanto en el colegio como en casa, pueden permitir que el niño con SA sea entrenado en sus necesidades como el aseo (programación horaria - entrenamiento) y el desarrollo de la capacidad de realizar por sí mismo las tareas cotidianas.
Fuentes:
- Angelman Syndrome Foundation.
- Asociación Síndrome de Angelman de Argentina.
- www.autismo.com/angel/informacion.htm
- Padres de Síndrome de Angelman México
Otras notas
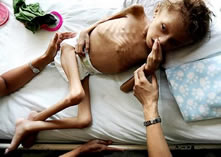
Es indiscutible la interrelación que existe entre pobreza y desnutrición, la cual ha motivado trabajos de investigación de diversos organismos. La desnutrición infantil es un síndrome clínico, caracterizado por un insuficiente aporte de proteínas y/o calo
Un estudio de Australia sugiere que las personas tratadas en centros especializados por traumatismos graves, sobrevivirían con menos discapacidades que los pacientes atendidos en otros hospitales.

El ministro de Salud de la Nación, Juan Manzur presentó ante representantes de sociedades científicas un plan para la detección temprana y el tratamiento...